
What is ATTR-CM?
Transthyretin amyloid cardiomyopathy (ATTR-CM) is a heart condition resulting from the buildup of an abnormal protein called transthyretin (TTR). In cases of ATTR-CM, the TTR protein misfolds and accumulates into structures known as amyloid fibrils. These fibrils then gather in the heart muscle and other body tissues. Over time, the accumulation of amyloid fibrils in heart muscle damages the muscle itself.1,2
In healthy people, TTR acts as a transport protein. It moves other molecules, such as the thyroid hormone thyroxine (T4) and retinol (Vitamin A), throughout the blood and cerebrospinal fluid.3 TTR misfolds due to aging or because of variants in the TTR gene. Such variants can be inherited, while others occur spontaneously.1
As amyloid fibrils collect inside the heart muscle, the muscle stiffens. This interferes with the heart’s ability to fill up with and pump blood normally. ATTR-CM is a progressive condition, meaning that it worsens over time. Eventually, it leads to heart failure.1,2,4
There are two types of ATTR-CM:
- Wild-type (wtATTR): The most common form of ATTR-CM, wild-type, occurs randomly. There’s no variant in the TTR gene, and it doesn’t run in families. Wild-type ATTR-CM usually develops as people get older and primarily affects white men over the age of 60, although women may also be affected.4,5
- Hereditary (hATTR): This type of ATTR-CM runs in families due to changes in the TTR gene that are passed down through generations.1 It affects men and women. Hereditary ATTR-CM may also cause protein buildup in the nerves, kidneys, and other organs.1 More than 120 gene mutations are associated with hATTR, with some variants affecting certain people more than others.6 For example, the ATTR variant V1221 mostly affects African Americans.6 However, simply having a TTR gene mutation doesn’t necessarily mean a person will develop ATTR-CM.
For many, arriving at a correct diagnosis can be challenging. Symptoms of ATTR-CM may mimic other medical conditions. In many cases, people experience nonspecific symptoms more often seen with general heart failure, such as fatigue, shortness of breath, or swelling in the legs.1
Additionally, while disease awareness among healthcare providers has improved, many providers still have difficulty recognizing ATTR-CM symptoms.3 Many people living with ATTR-CM experience delays in receiving a correct diagnosis, even after symptoms begin. Sometimes these delays can last years. Unfortunately, this means ATTR-CM is usually advanced, and symptoms may already be severe, at the time of diagnosis.1
Prevalence of ATTR-CM
Because this condition is often underdiagnosed, the exact prevalence of ATTR-CM is unknown. However, based on available data, it’s a rare and underrecognized cause of heart failure.2
Causes and Risk Factors
What Causes ATTR-CM?
TTR amyloid cardiomyopathy occurs when a specific protein, known as TTR, folds abnormally. This leads to the formation of amyloid fibrils, which can accumulate inside the heart muscle.6
A portion of the heart muscle, known as the myocardium, is one of the most common sites of accumulation of misfolded TTR proteins. Over time, the heart muscle stiffens as a result of this buildup. Ultimately, this results in heart failure.4
ATTR-CM and Genetics
In cases of wild-type ATTR-CM, it’s believed that the normal aging process contributes to the TTR protein misfolding, even if the gene that codes for TTR is normal.7
With hereditary ATTR-CM, structural changes to the TTR protein result from genetic mutations in the gene that codes for TTR. This genetic mutation runs in families — there are more than 120 identified mutations that may affect the TTR gene. The most common is the V1221 mutation, which almost exclusively affects African Americans.6,7
ATTR-CM Prevention and Screening
Currently, there’s no way to prevent ATTR-CM.7 However, genetic testing and counseling may be available for people with a family history of the disease.7 Additionally, the increased use of artificial intelligence (AI) algorithms may help doctors screen for the condition, making it an important tool to help identify patients at risk for ATTR-CM.8
It’s also important to be aware of some of the more common symptoms of ATTR-CM, which may include:1
- Shortness of breath
- Chest congestion
- Fatigue
- Swelling in the lower extremities
- Abnormal heart rhythms
- Carpal tunnel syndrome
Types of ATTR-CM
There are two types of ATTR-CM:
Occurs randomly and may be influenced by changes occurring during the normal aging process.1
Runs in families and results from mutations in the TTR gene.1,6
Symptoms of ATTR-CM
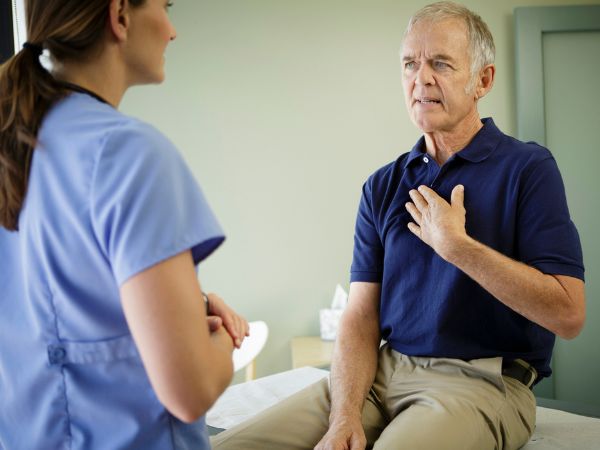
Some people don’t have symptoms until their condition is advanced.4 However, there are several more common “red flag symptoms” associated with ATTR-CM, such as:1,4,6
- Fatigue
- Shortness of breath
- Swelling in the lower legs
- Heart failure
- Heart rhythm abnormalities like atrial fibrillation
- Carpal tunnel syndrome
- Lumbar spinal stenosis
- Digestive problems
ATTR-CM Complications
When left untreated, ATTR-CM may lead to worsening heart failure, arrhythmias, and conduction system issues. These, in turn, may cause complete heart block or sudden cardiac death.8
Diagnosis and Treatment
ATTR-CM Diagnosis
An accurate diagnosis of ATTR-CM usually comes after doctors take a complete history and perform a physical examination. These procedures are followed by routine cardiac tests, usually an electrocardiogram (EKG) and echocardiogram.1,6,10
To confirm a diagnosis, further testing is usually necessary. Doctors may recommend blood tests to rule out AL amyloidosis, another form of amyloidosis that’s unrelated to TTR.2 In some cases, a nuclear scan can help confirm TTR amyloidosis in the heart muscle — other people require a heart muscle tissue biopsy using Congo red staining to confirm the presence of amyloid deposits.2,6,8
For some people, genetic testing for mutations that cause ATTR-CM can be helpful, especially if there’s a family history of the condition.
ATTR-CM Treatment
The treatment of ATTR-CM, regardless of type, focuses on slowing down the deposits of misfolded TTR while also managing symptoms. Since many ATTR-CM symptoms mimic those of heart failure, lifestyle interventions can be beneficial. These may include:
- Dietary sodium restriction2
- Use of compression stockings as necessary2
- Monitoring fluid intake11
- Regular physical activity11
Treatments targeting the TTR protein itself may include TTR stabilizers, which help prevent the TTR protein from misfolding. Or doctors may suggest a TTR gene silencer, which targets TTR messenger RNA. These gene silencers suppress levels of circulating TTR protein in the body.12
Frequently Asked Questions About ATTR-CM
- What is ATTR-CM?
TTR amyloid cardiomyopathy (ATTR-CM) is a heart condition resulting from the buildup of amyloid fibrils in the heart muscle tissue. These amyloid fibrils develop due to a misfolded protein called TTR.1
In healthy people, TTR helps move other molecules, such as the thyroid hormone thyroxine (T4) and Vitamin A, throughout the body.2 TTR becomes unstable as a person gets older, or because of variants in the TTR gene, which may be inherited or occur randomly.1
As amyloid fibrils accumulate in the heart muscle, the muscle stiffens and cannot normally fill with and pump blood. ATTR-CM is a progressive condition that worsens over time, eventually leading to heart failure.1,3
- Are there different types of ATTR-CM?
There are two different types of ATTR-CM, wild-type and hereditary.
- Wild-type ATTR-CM is associated with aging. It occurs randomly and there’s no variant in the TTR gene, meaning it does not run in families.1
- Hereditary ATTR-CM runs in families, due to changes in the TTR gene.1
- What are some common clinical signs and symptoms of ATTR-CM?
- Some common ATTR-CM symptoms are like those of heart failure and include:1,2,3,4
- Fatigue
- Shortness of breath
- Swelling in the lower legs
- Heart rhythm abnormalities
- Other symptoms that may appear unrelated to the heart include but are not limited to:8
- Numbness or tingling in the hands or feet
- Carpal tunnel syndrome
- Lumbar spinal stenosis
- GI issues
- Some common ATTR-CM symptoms are like those of heart failure and include:1,2,3,4
- What treatments are available for ATTR-CM?
The treatment of ATTR-CM focuses on managing a person’s symptoms and slowing the accumulation of misfolded TTR.1 Approved therapies for ATTR-CM include TTR stabilizers and TTR gene silencers.8,13 The goal of treatment is to slow disease progression, which is why early diagnosis is so important.
- If I’m diagnosed, where do I begin?
If you have a confirmed diagnosis of ATTR-CM, it’s important to speak with your doctor about approved treatment options to find the most appropriate treatment plan. There are also amyloidosis patient support groups that can provide helpful information.
- How do I get screened for ATTR-CM?
If your doctor suspects ATTR-CM, there are a number of tests they may recommend. These may include blood and urine tests, an electrocardiogram (ECG), an echocardiogram and a nuclear scan called a scintigraphy test. These tests help to rule out other forms of amyloidosis and to detect amyloid fibrils in heart tissue.2
Learn More About ATTR-CM
Explore ATTR-CM clinical trials at ClinicalTrials.gov.
- References
- Transthyretin amyloid cardiomyopathy (ATTR-CM). American Heart Association. Updated May 29, 2024. Accessed February 13, 2026. https://www.heart.org/en/health-topics/cardiomyopathy/what-is-cardiomyopathy-in-adults/transthyretin-amyloid-cardiomyopathy-attr-cm.
- Transthyretin-mediated amyloid cardiomyopathy (ATTR-CM). Rare Disease Advisor. Updated November 26, 2024. Accessed March 31, 2026. https://www.rarediseaseadvisor.com/disease-info-pages/transthyretin-mediated-amyloid-cardiomyopathy-overview/.
- Zhang KW, Stockerl-Goldstein KE, Lenihan DJ. Emerging therapeutics for the treatment of light chain and transthyretin amyloidosis. JACC: Basic to Translational Science. 2019;4(3):438-448.
- Cardiac amyloidosis. World Heart Federation. Accessed February 13, 2026. https://world-heart-federation.org/world-heart-day/cvd-causes-conditions/cardiac-amyloidosis/.
- Aimo A, Panichella G, Garofalo M, et al. Sex differences in transthyretin cardiac amyloidosis. Heart Failure Reviews. 2023;29(2):321.
- Hereditary ATTR amyloidosis. Amyloidosis Research Consortium. Accessed February 13, 2026. https://arci.org/about-amyloidosis/hereditary-attr-amyloidosis/.
- Genetic testing and counseling for hereditary transthyretin amyloidosis (hATTR). American Heart Association. https://www.heart.org/en/health-topics/cardiomyopathy/understand-your-risk-for-cardiomyopathy/genetic-testing-for-hattr-amyloidosis. Updated May 31, 2024. Accessed June 15, 2026.
- Jain A, Zahra F. Transthyretin amyloid cardiomyopathy (ATTR-CM). In: StatPearls. StatPearls Publishing; 2025. Updated April 27, 2023. Accessed February 13, 2026. https://www.ncbi.nlm.nih.gov/books/NBK574531/.
- Harnessing the power of AI to detect ATTR-CM. Pfizer.com. https://www.pfizer.com/news/articles/harnessing_the_power_of_ai_to_detect_attr_cm. Accessed May 29, 2026.
- Porcari A, Sinagra G, Gillmore JD, Fontana M, Hawkins PN. Breakthrough advances enhancing care in ATTR amyloid cardiomyopathy. European Journal of Internal Medicine. 2024;123:29-36.
- Living with transthyretin amyloid cardiomyopathy: A toolkit for patients. World Heart Federation. February 2025. Accessed February 13, 2026. https://world-heart-federation.org/wp-content/uploads/WHF_ATTR-Tool-Kit.pdf.
- Tschöpe C, Elsanhoury A. Treatment of transthyretin amyloid cardiomyopathy: the current options, the future, and the challenges. Journal of Clinical Medicine. 2022;11(8):2148.
- BritoD, Albrecht FC, Arenaza DP de, et al. World heart federation consensus on transthyretin amyloidosis cardiomyopathy (Attr-cm). Global Heart. 2023;18(1):59.
The information contained on this page is provided for your general information only. It is not intended as a substitute for seeking medical advice from a healthcare provider. Pfizer is not in the business of providing medical advice and does not engage in the practice of medicine. Pfizer under no circumstances recommends particular treatments for specific individuals and in all cases recommends consulting a physician or healthcare center before pursuing any course of treatment.