How Does It Feel to Have Sickle-Cell Disease?: When Listening to Patients Gives Researchers Clues
Molecular biologists certainly have the insider’s view of a disease. They know (or try to find out) how it operates in the body on a cellular level. But the mechanisms of a particular condition aren’t the only thing that matters to these scientists. Starting “outside,” say by learning more about what it’s like to have a disease, and how people generally cope with symptoms such as pain, can help a researcher develop treatments that truly address patients’ needs.
Debra Pittman, a research fellow at Pfizer, wasn’t testing medicines when she and colleagues began their six-month longitudinal study of 35 patients with sickle-cell disease. Instead, the team planned to collect blood samples and other biological markers of health, while also tracking patients’ subjective experiences of their disease – a particularly painful and debilitating one that affects approximately 100,000 Americans.
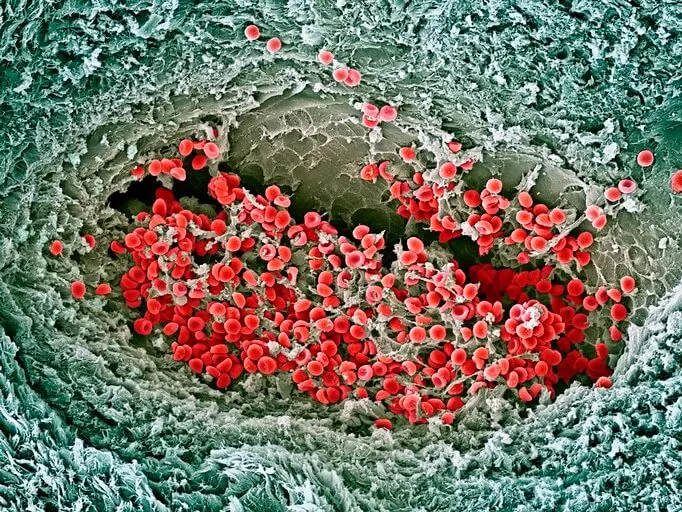
Sickle-cell disease distorts red blood cells into “sickle” or crescent shapes. When these stiff, misshaped cells get stuck and block small blood vessels, depriving tissues and organs of oxygen-rich blood, it can lead to what’s called a vaso-occlusive crisis. This crisis causes severe pain and fatigue, Pittman says, and can lead to organ failure, stroke and other problems.
“We don’t know what precipitates a crisis,” Pittman says. “Are there any biomarkers, such as the numbers of certain cells that show up in blood tests, associated with it? Can we start to define it differently, and how does the patient think about this? What do they do when they’re having a crisis?”
In an attempt to answer those questions, Pittman and colleagues at Pfizer, Wayne State University of Medicine and Dana-Farber/Boston’s Children tracked patients in Detroit, Michigan. Sanguine Biosciences provided mobile phlebotomy and traveled to the patients’ homes for regular blood draws.
The team developed an electronic patient-reported outcomes tool that enabled patients to record their fatigue, pain levels and prescription pain-killer drug use. If they thought they were having a vaso-occlusive crisis, patients could push a button to summon the phlebotomist to draw blood within 24 hours. Their blood would then be drawn again after 48 and 72 hours passed. “We were trying to capture any changes during and after the crises,” Pittman says.
The study found that the majority of patients had at least one to three vaso-occlusive events over the six-month period. Most eye-opening was the fact that in 63 percent of such cases, the patient did not seek any kind of medical intervention, even though general results showed that significantly different pain, functioning and fatigue levels were reported during these crises.
“If 63 percent of patients are being treated at home, but the clinical criteria for a vaso-occlusive crisis is a hospitalization greater than four hours, it makes it hard to do clinical trials,” says Pittman. In other words, the way researchers have defined a key outcome in sickle-cell disease studies is perhaps not reflective of the true state of many patients. It’s a highly practical insight gleaned from simply asking a group of people, in a structured way, how they cope with their illness.
Listening carefully to patients adds value to whatever tests might be underway in the lab. More importantly, it enhances empathy, a deep feeling of concern for others’ suffering that motivates scientists like Pittman to keep pushing for the next breakthrough. “Sickle-cell disease has such a dramatic effect on people’s lives.”