How Quantum Physics and AI is Disrupting Drug Discovery & Development
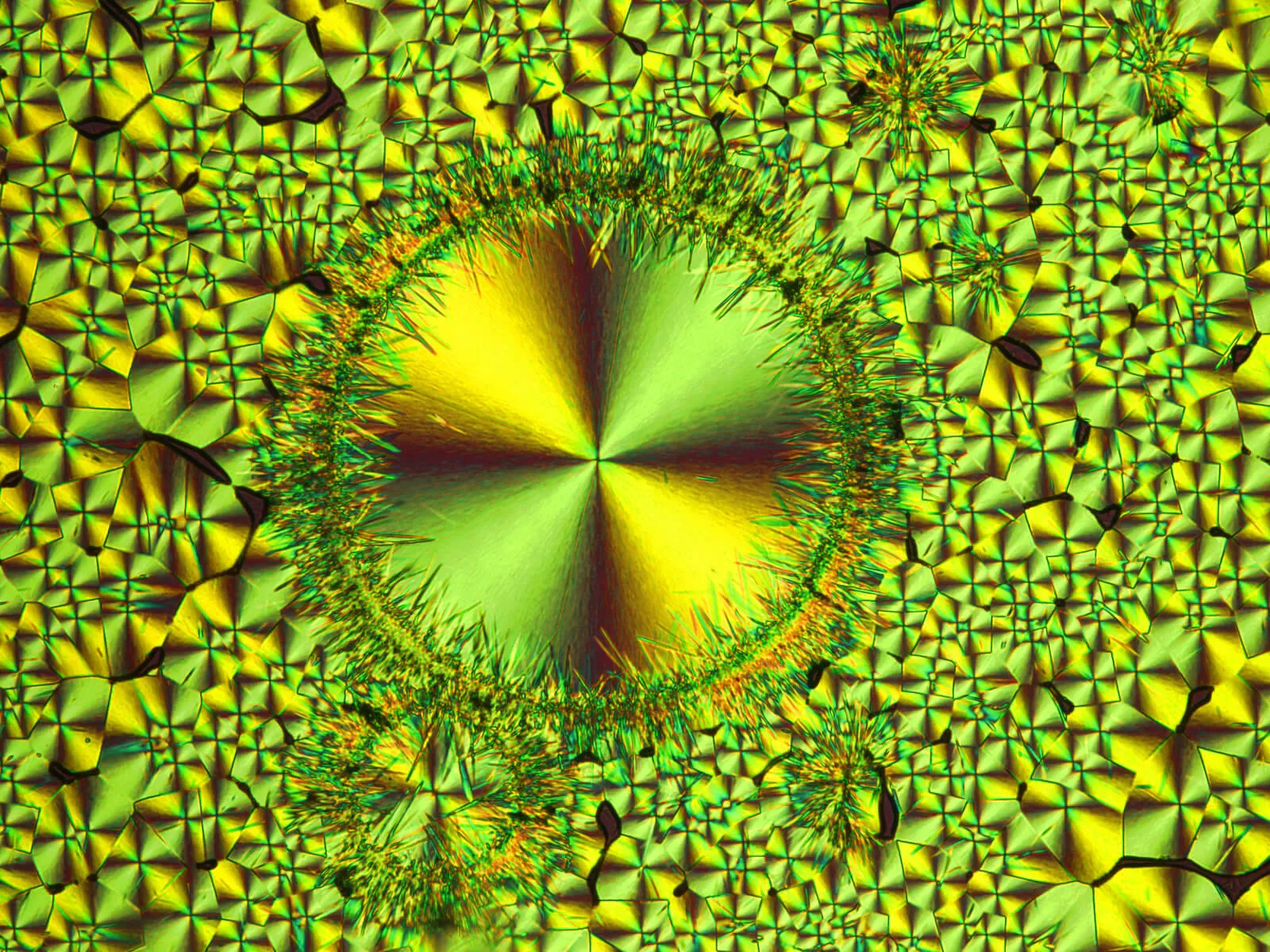
When a new molecule is a potential drug candidate, scientists want to learn as much about the molecule as possible; its shape, size and other properties down to the electron level. To do this they have traditionally used a technique called X-ray crystallography. In a multi-step, time-consuming process the compound is converted into crystal form and then an X-ray beam is shot through it to determine its 3-D structure.
In recent years, scientists have begun using a computer modelling technique known as crystal structure prediction (CSP) to do virtual crystallography. By applying quantum physics, scientists can predict the behavior of electrons in a molecule to determine its 3-D structure. CSP involves very many complicated mathematical calculations that take up a lot of computing power and up to four months.
But now thanks to a recent strategic research collaboration with XtalPi, a U.S.-China pharmaceutical tech company, Pfizer scientists are performing these calculations in a matter of days. Pioneered by a group of quantum physicists from MIT, the XtalPi technology leverages artificial intelligence and cloud computing to perform these complex equations. “A process that used to take us so much time that we almost didn’t event attempt it has become the norm, and now we can try it on almost every small molecule project,” says Bruno Hancock, Global Head of Materials Science at Pfizer’s Groton, Connecticut research site.
As one of the early collaborators with XtalPi, Pfizer, bringing its deep experience in the field, helped establish new techniques for early drug screening. “This collaboration is already changing the way Pfizer performs its screening work and has the potential to disrupt the industry as a whole,” says Geoff Wood, a Principal Scientist, also based at Pfizer’s Groton site.
Crowding the Servers
While electrons are one of the smallest pieces of matter, predicting their movement involves a massive amount of computational power. To perform a single crystal structure prediction requires the computing power equivalent to one million laptops, says Hancock. “When they (cloud provider) do one of these calculations for us, XtalPi becomes the single biggest user of those services in the USA at that moment in time,” says Hancock.

At the microscale, what they’re calculating is the properties of electrons in a molecule. But since molecules have many electrons that are constantly moving and changing, they must perform multiple calculations simultaneously. “It can take billions of calculations to come up with a final answer,” says Hancock.
Narrowing down to the best one
Once scientists can calculate the predicted 3-D structure of a drug molecule, they can use it to predict its mechanical and chemical properties, such as its shape, solubility and melting point, and how it binds with a protein receptor.
In this way, scientists are selecting what they predict will be optimal molecule based on theoretical predictions, rather than traditional experimental methods. “There are hundreds of thousands of molecules that we’re considering, and we want the best one,” says Hancock. “One way to find the best compound is to make a large number compounds and measure their properties, which is time and resource intensive. Or the other way is to predict what each compound’s properties are going to be, and narrow down to what are predicted to be the best ones. Using the approaches together we have greater effectiveness in small molecule drug discovery and development.”




